En las guías existe consenso en que la mayoría de los tumores suprarrenales (tumores de la glándula suprarrenal) deben operarse de forma mínimamente invasiva cuando está indicada la cirugía. Un diámetro tumoral > 6 cm y claros indicios de malignidad en la imagen preoperatoria se consideran límites de la cirugía mínimamente invasiva. En cuanto al tamaño del tumor, se suele aplicar la regla de los 6 cm, y los tumores más grandes deben extirparse mediante cirugía abierta convencional. Sin embargo, numerosos estudios han demostrado que, incluso en tumores suprarrenales grandes > 6 cm, es posible proceder de forma mínimamente invasiva segura con la experiencia adecuada. Debido a la falta de evidencia, la regla de los 6 cm no puede considerarse un límite absoluto. No obstante, en general se recomienda la adrenalectomía convencional en tumores grandes, especialmente cuando hay sospecha de malignidad en las imágenes.
Los procedimientos laparoscópicos y retroperitoneoscópicos son equivalentes. La elección de la vía de acceso depende de la experiencia y la preferencia del cirujano.
Bemerkung: La adrenalectomía laparoscópica (AE) no ha cambiado sustancialmente desde su primera descripción en 1992 y es el método preferido en muchos lugares debido al acceso y la orientación familiares.
La resección parcial suprarrenal (adrenalectomía preservadora de la glándula suprarrenal) tiene un papel en pacientes con síndrome de Conn causado por tumores pequeños, generalmente excéntricos, y en tumores bilaterales en los que se busca preservar la función adrenocortical. En estos casos debe conservarse al menos un tercio de una glándula suprarrenal. No es necesario conservar la vena suprarrenal central. En caso de cirugía unilateral, la producción de hormonas corticoides suele ser asumida completamente por la glándula suprarrenal contralateral.
La indicación de adrenalectomía mínimamente invasiva está dada en las siguientes situaciones:
- Tumores endocrinamente activos de la corteza suprarrenal (adenomas de Conn o de Cushing, tumores con secreción de hormonas sexuales) de hasta 10 cm
- Síndrome de Conn (hiperaldosteronismo primario, PHA):
La causa más frecuente de hipertensión secundaria es el hiperaldosteronismo primario.
Cribado en incidentaloma con hipertensión simultánea y/o hipopotasemia poco clara. Morfológicamente puede haber hiperplasia suprarrenal o uno o varios adenomas.
En caso de adenoma productor de aldosterona unilateral o hiperplasia suprarrenal unilateral está indicada la adrenalectomía unilateral. En el adenoma productor de aldosterona solitario también puede realizarse una extirpación parcial de la glándula suprarrenal afectada.
En pacientes con hiperaldosteronismo primario (PHA) y alteraciones suprarrenales bilaterales puede considerarse una adrenalectomía unilateral si el muestreo venoso suprarrenal (AVS; extracción selectiva de sangre de las venas suprarrenales) demuestra una lateralización funcional clara. La importancia de la PET con CXCR4 como alternativa no invasiva al AVS debe validarse con más estudios.
- Síndrome de Cushing(hipercortisolismo)
Un síndrome de Cushing adrenal florido con estigmas clínicos clásicos constituye una indicación quirúrgica.
Una situación especial se presenta en la secreción autónoma leve de cortisol (MACS). Por definición, los pacientes con MACS no presentan signos clínicos de síndrome de Cushing, pero sí una prueba de supresión con dexametasona de 1 mg patológica.
Una situación especial se presenta en la secreción autónoma leve de cortisol (MACS). Por definición, los pacientes con MACS no presentan signos clínicos de síndrome de Cushing, pero sí una prueba de supresión con dexametasona de 1 mg patológica.
Un gran estudio publicado en 2022 mostró que los pacientes con secreción autónoma de cortisol presentan una mayor mortalidad global en comparación con las personas con adenoma hormonalmente inactivo (Deutschbein et al., Lancet Diabetes & Endocrinology, 2022). Según la guía revisada de incidentalomas de la European Society of Endocrinology de 2023, en la MACS debe considerarse siempre un tratamiento quirúrgico, especialmente si hay comorbilidades relevantes.
Antes de una posible extirpación quirúrgica del tumor suprarrenal debe confirmarse la independencia de ACTH del exceso de cortisol, para que la intervención no se realice erróneamente cuando la causa del exceso hormonal es, por ejemplo, hipofisaria por un adenoma de lóbulo anterior de la hipófisis (síndrome de Cushing central) o un síndrome paraneoplásico con secreción ectópica de ACTH en una enfermedad tumoral.
- Tumores de la corteza suprarrenal productores de hormonas sexuales
Un carcinoma adrenocortical es la causa más frecuente de secreción patológica clínicamente relevante de andrógenos/estrógenos desde la glándula suprarrenal; los adenomas son muy raros.
- Síndrome de Conn (hiperaldosteronismo primario, PHA):
- Carcinoma de corteza suprarrenal Carcinoma adrenocortical (ACC) hasta el estadio II de ENSAT
Enlace a la clasificación ENSAT y tamaño ≤ 6 cm- En el momento del diagnóstico casi siempre > 4 cm y presenta actividad endocrina en el 50-80 % de los casos. Es típica la producción de cortisol o una producción hormonal mixta (andrógenos/estrógenos y cortisol).
- En el carcinoma de corteza suprarrenal la adrenalectomía abierta es el estándar de oro. En tumores < 6 cm sin indicios de infiltración local o ganglionar (estadio ENSAT I+II) puede realizarse una adrenalectomía mínimamente invasiva.
- Se recomienda una resección tumoral radical con extirpación de la glándula suprarrenal y de todo el tejido adiposo/conectivo del compartimento afectado sin apertura de la cápsula, junto con linfadenectomía (esta última con datos inciertos). Hasta la fecha no existe una definición sobre la extensión necesaria de la linfadenectomía.
- Bemerkung: Según la evidencia actual, la tasa de recidivas locales y peritoneales está aumentada en el grupo laparoscópico. La conversión de adrenalectomía laparoscópica a abierta empeora la supervivencia global.
- Feocromocitoma (PC) Tumor de la médula suprarrenal con exceso de catecolaminas
- Aproximadamente un tercio de todos los pacientes con feocromocitoma pertenecen a un síndrome tumoral hereditario. El cribado genético es un componente indispensable del diagnóstico de feocromocitoma.
- Las formas sindrómicas familiares incluyen la neoplasia endocrina múltiple tipo 2 (MEN 2), el síndrome de von Hippel-Lindau (VHL), la neurofibromatosis tipo 1 (NF1) y las mutaciones germinales de la subunidad B y D de la succinato deshidrogenasa (SDHB y SDHD).
- Se diferencian de las formas esporádicas por la edad de presentación, la localización tumoral y el riesgo de malignización.
- Los feocromocitomas hereditarios asociados a síndromes tienen una tasa de malignidad más baja que los tumores esporádicos, pero a menudo son multifocales/bilaterales.
- Una excepción importante son los tumores asociados a SDHB, que presentan un riesgo especialmente alto de malignidad y recidiva.
- Las resecciones preservadoras de órgano deben preferirse cuando esté indicada (tumores hereditarios, hallazgos bilaterales, pacientes jóvenes), siempre que sea oncológicamente seguro, para evitar una insuficiencia suprarrenal postoperatoria y reducir la necesidad de dependencia de esteroides de por vida.
- En total, el 10 % de los PC son malignos. La única prueba de un feocromocitoma maligno son las metástasis.
- Incidentalomas
| Tamaño del incidentaloma | Recomendación |
|---|---|
| < 4 cm | No cirugía si es hormonalmente inactivo y la imagen es benigna |
| 4 – 6 cm | Decisión individual («zona gris»), valoración del riesgo de malignidad, imagen, crecimiento, deseo del paciente, re-TC/RM en 6-12 meses |
| > 6 cm | Cirugía recomendada, incluso en tumores hormonalmente inactivos por mayor riesgo de carcinoma (25 %) |
Un incidentaloma suprarrenal es un hallazgo casual, generalmente asintomático, de una masa (RF) en la glándula suprarrenal que no se descubrió por sospecha clínica de enfermedad endocrina. Habitualmente se considera relevante un tamaño a partir de 1 cm de diámetro.
Debido a la posible malignidad y actividad hormonal, todo incidentaloma suprarrenal a partir de 1 cm de tamaño debe estudiarse de forma adecuada según las guías mediante métodos bioquímicos y radiológicos.
Además del tamaño, debe evaluarse la probabilidad de un tumor maligno. Para ello, la guía recomienda en primer lugar una tomografía computarizada (TC) sin contraste con determinación de unidades Hounsfield (HU) para medir la densidad de los tumores. En una masa homogénea con densidad ≤ 10 HU no se requiere más imagen. En tumores homogéneos pequeños de hasta 4 cm de diámetro y HU entre 11 y 20 debe realizarse imagen adicional, por ejemplo mediante resonancia magnética (RM) con «chemical-shift», TC con «wash-out» o tomografía por emisión de positrones con fluorodesoxiglucosa (véase Diagnóstico más abajo). Los tumores más grandes, heterogéneos o con HU > 20 deben discutirse en una reunión interdisciplinaria. A continuación debe realizarse imagen adicional rápida o evaluarse la cirugía. Si tras imagen ampliada y discusión interdisciplinaria no se establece indicación quirúrgica, se recomienda imagen de seguimiento a los 6-12 meses.
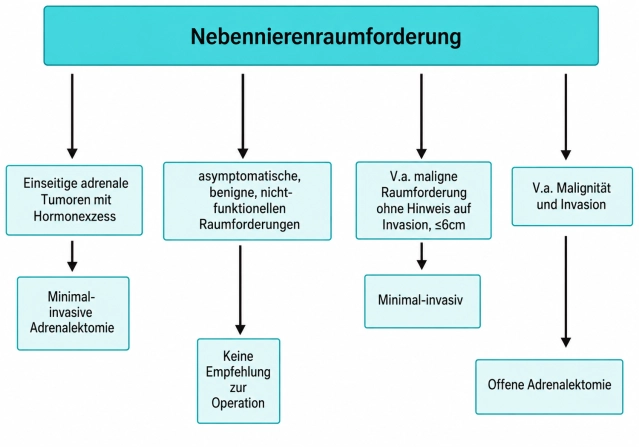
- Metástasis suprarrenales (de tumores malignos de otro origen) deben extirparse si no hay otras metástasis y la extirpación consigue la ausencia de tumor. La adrenalectomía de metástasis se realiza de forma mínimamente invasiva siempre que la metástasis pueda extirparse en bloque y sin diseminación de células tumorales. El abordaje abierto queda reservado a los pocos casos en los que hay indicios de infiltración local o cuando la metástasis supera los 6 cm.
- Mielolipomas/quistes suprarrenales no constituyen indicación quirúrgica. El diámetro tumoral no tiene valor pronóstico, ya que son tumores benignos. El dolor en el flanco causado por el tamaño del tumor o una hemorragia en el retroperitoneo puede constituir rara vez indicación quirúrgica.
Disección ganglionar
En caso de ganglios linfáticos aumentados de tamaño debe realizarse una linfadenectomía locorregional. Los datos sobre la linfadenectomía periadrenal/renal-hiliar son insatisfactorios.
