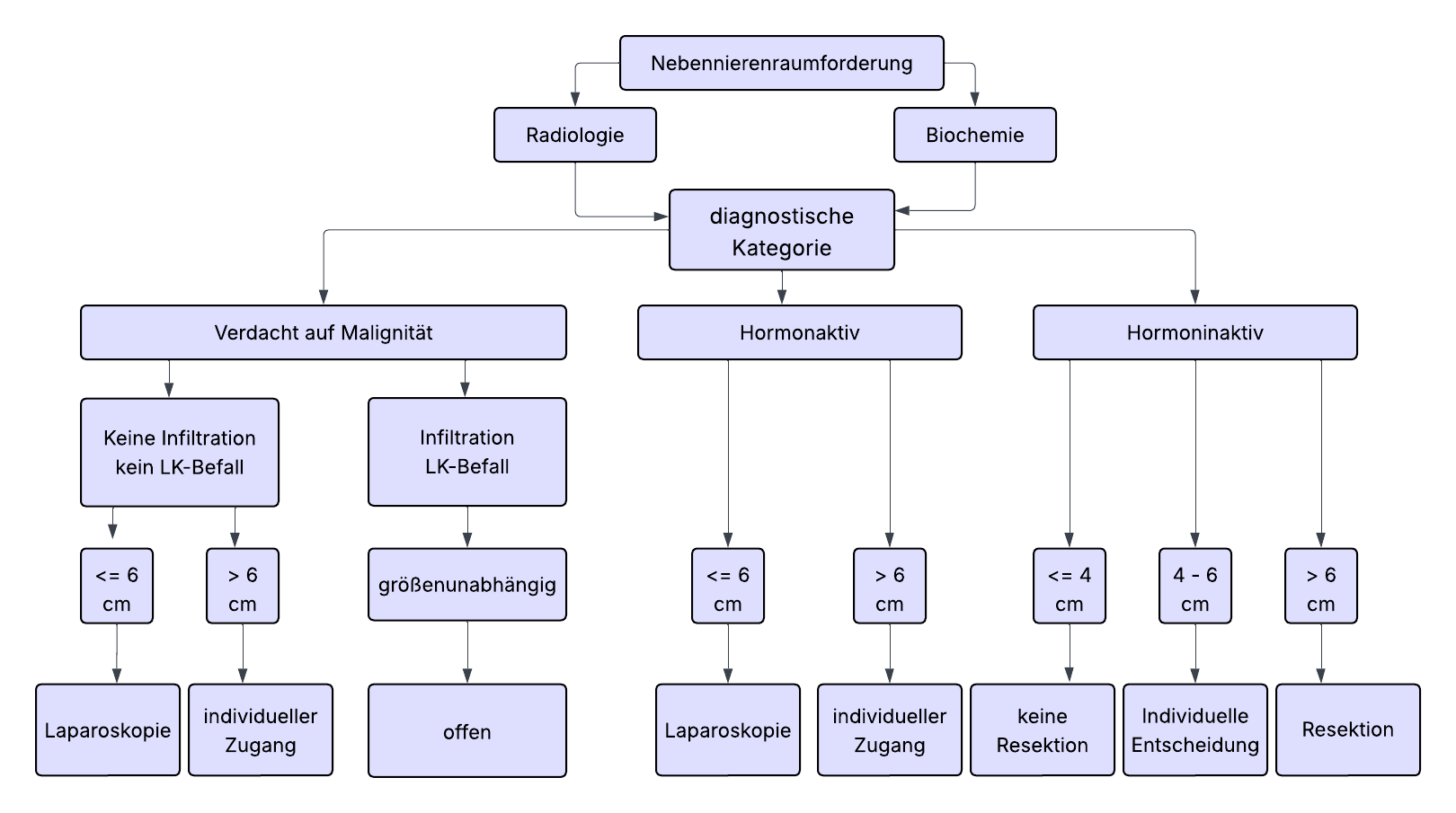
Há consenso nas diretrizes de que a maioria dos tumores adrenais (TAs) deve ser operada de forma minimamente invasiva, se indicado. Diâmetro do tumor > 6 cm e evidência clara de malignidade na imagem pré-operatória são considerados limites da cirurgia minimamente invasiva. Em relação ao tamanho do tumor, uma regra de 6 cm é frequentemente assumida, tumores maiores devem ser excisados de forma convencional aberta. Em contraste, numerosos estudos mostraram que mesmo tumores adrenais grandes > 6 cm podem ser abordados de forma minimamente invasiva com segurança, com expertise apropriada. Devido à falta de evidências, a regra de 6 cm não pode, portanto, ser considerada um limite absoluto. Essa decisão requer informações especiais e consentimento dos pacientes. Se isso estiver presente, a adrenalectomia, se o tumor puder ser completamente removido sem ruptura da cápsula, pode ser realizada de forma minimamente invasiva, independentemente do tamanho do tumor.
No entanto, a adrenalectomia convencional é geralmente recomendada para tumores grandes, especialmente em casos de suspeita de malignidade na imagem.
Procedimentos laparoscópicos e retroperitoneoscópicos são equivalentes. A escolha da via de acesso depende da experiência e preferência do cirurgião.
Nota: A adrenalectomia laparoscópica (AE) não mudou tecnicamente de forma significativa desde sua primeira descrição em 1992 e é o método preferido em muitos lugares devido ao acesso e orientação familiares.
A ressecção adrenal parcial (adrenalectomia poupadora de adrenal) tem um papel em pacientes com síndrome de Conn, que é causada por tumores pequenos, geralmente localizados excentricamente, e tumores bilaterais, onde a intervenção cirúrgica visa preservar a função adrenocortical. O objetivo é preservar pelo menos um terço de uma glândula adrenal. A veia adrenal central não precisa ser preservada. Em cirurgia unilateral apenas, a produção de hormônio corticosteróide é geralmente completamente assumida pela glândula adrenal contralateral.
- Tumores endócrino-ativos do córtex adrenal (adenomas de Conn ou Cushing, tumores com secreção de hormônios sexuais) até 10 cm
- Síndrome de Conn (hiperaldosteronismo primário, HAP):
A causa mais comum de hipertensão secundária é o hiperaldosteronismo primário.
Rastreamento para incidentaloma e hipertensão concomitante e/ou hipocalemia inexplicada. Morfologicamente, pode haver hiperplasia adrenal ou um ou mais adenomas.
Em adenoma produtor de aldosterona unilateral ou hiperplasia adrenal unilateral, a adrenalectomia unilateral é indicada. Em adenoma produtor de aldosterona solitário, a remoção parcial da glândula adrenal afetada também pode ser realizada.
Em pacientes com HAP e alterações adrenais bilaterais, a cirurgia pode ser considerada se a amostragem venosa adrenal (AVS por meio de amostragem seletiva de sangue da veia adrenal) mostrar localização funcional.
- Síndrome de Cushing(hipercortisolismo)
Uma síndrome de Cushing adrenal florida com estigmas clínicos clássicos representa uma indicação para cirurgia.
Na síndrome de Cushing subclínica, há apenas uma indicação relativa para cirurgia. Aqui, uma pessoa afetada mostra excesso bioquímico de cortisol, mas a manifestação clínica está ausente. Dados comparativos sobre mortalidade ou eventos cardiovasculares de cirurgia versus terapia médica de comorbidades estão faltando. A decisão pela cirurgia depende principalmente da idade e desejos do paciente.
Antes da possível remoção cirúrgica do TA, a independência de ACTH do excesso de cortisol deve ser confirmada para que o procedimento não seja realizado erroneamente, embora a causa do excesso hormonal seja, por exemplo, pituitária.
Exclusão de hipercortisolismo dependente de ACTH (adenoma do lobo anterior da pituitária) (síndrome de Cushing central) ou síndrome paraneoplásica com secreção ectópica de ACTH em doença tumoral.
- Tumores do córtex adrenal produtores de hormônios sexuais
Um carcinoma adrenocortical é a causa mais comum de secreção androgênica/estrogênica patológica clinicamente relevante da glândula adrenal, adenomas são muito raros.
- Síndrome de Conn (hiperaldosteronismo primário, HAP):
- Carcinoma do córtex adrenal Carcinoma adrenocortical (CAC) até o estágio II do ENSAT
Link para a classificação ENSAT e tamanho ≤ 6cm- No diagnóstico inicial, quase sempre > 4cm e mostra atividade endócrina em 50-80%. Tipicamente, há produção de cortisol ou produção hormonal mista (andrógenos/estrógenos e cortisol).
- Para carcinoma do córtex adrenal, a adrenalectomia aberta é o padrão ouro. Para tumores < 6 cm sem evidência de infiltração local ou linfonodal (ENSAT std. I+II), a adrenalectomia minimamente invasiva pode ser realizada.
- Uma ressecção tumoral radical com remoção da glândula adrenal e todo o tecido adiposo/conjuntivo no compartimento afetado sem abertura da cápsula com linfadenectomia, isso em situação de dados incertos, é recomendada. Uma definição quanto à extensão da linfadenectomia necessária ainda não existe.
- Nota: A taxa de recidivas locais e peritoneais é aumentada no grupo laparoscópico de acordo com as evidências atuais. A conversão de laparoscópica para adrenalectomia aberta piora a sobrevida global.
- Feocromocitoma (FC) Tumor da medula adrenal com excesso de catecolaminas
- Cerca de 1/3 de todos os pacientes com feocromocitoma são atribuíveis a uma síndrome tumoral hereditária. O rastreamento genético é uma parte indispensável do diagnóstico de feocromocitoma.
- Formas sindrômicas familiares incluem neoplasia endócrina múltipla tipo 2 (NEM 2), síndrome de von Hippel-Lindau (VHL), neurofibromatose tipo 1 (NF1) e mutações germinativas da subunidade B e D da succinato desidrogenase (SDHB e SDHD).
- Eles diferem das formas esporádicas na idade de início, localização do tumor e seu risco de degeneração maligna.
- Feocromocitomas hereditários associados a síndromes têm uma taxa de malignidade menor que tumores esporádicos, mas frequentemente ocorrem de forma multifocal/bilateral. O objetivo aqui é possibilitar uma vida sem dependência de esteroides. Portanto, nessa situação, uma ressecção adrenal minimamente invasiva poupadora de órgão deve ser discutida.
- Uma exceção importante são tumores associados a SDHB com malignidade e risco de recidiva particularmente altos.
- No geral, 10% dos FCs são malignos. A prova de um feocromocitoma maligno é exclusivamente metástases.
- Incidentalomas
| Tamanho do incidentaloma | Recomendação |
|---|---|
| < 4 cm | Nenhuma cirurgia se hormonalmente inativo e imagem benigna |
| 4 – 6 cm | Decisão individual ("área cinzenta"), pesando risco de malignidade, imagem, crescimento, preferência do paciente, TC/RM de repetição em 6 - 12 meses |
| > 6 cm | Cirurgia recomendada, mesmo para tumores hormonalmente inativos devido ao risco aumentado de carcinoma (25%) |
Um incidentaloma adrenal é uma massa (lesão) incidental, geralmente assintomática, da glândula adrenal que não foi descoberta devido a suspeita clínica de uma doença endócrina. Geralmente, um tamanho de 1 cm de diâmetro ou mais é considerado um achado relevante. Lesões menores são frequentemente consideradas achados incidentais sem necessidade de ação adicional, a menos que mostrem anormalidades.
Nota: Incidentalomas devem ser avaliados usando métodos bioquímicos e radiológicos.
Apenas infiltração detectável de estruturas vizinhas ou detecção de metástases distantes são prova de malignidade. A determinação de unidades Hounsfield (HU) pode fornecer pistas adicionais.
- Mielolipomas/cistos adrenais não representam uma indicação para cirurgia. O diâmetro do tumor é prognosticamente insignificante, pois esses são tumores benignos. Dor no flanco devido ao tamanho do tumor ou hemorragia no retroperitônio pode raramente representar uma indicação para cirurgia.
- Metástases adrenais (de tumores malignos de outras origens) devem ser removidas se não houver outras metástases e o status livre de tumor for alcançado pela remoção.
A adrenalectomia de metástase deve ser realizada de forma minimamente invasiva, desde que a metástase possa ser removida in toto e sem semeadura de células tumorais. Uma abordagem aberta permanece reservada para os poucos casos onde há evidência de infiltração local ou se a metástase exceder 6 cm.
Dissecção de linfonodos
Em linfonodos aumentados, a linfadenectomia locorregional deve ser realizada. A situação de dados quanto à linfadenectomia periadrenal/hilar renal é insatisfatória.