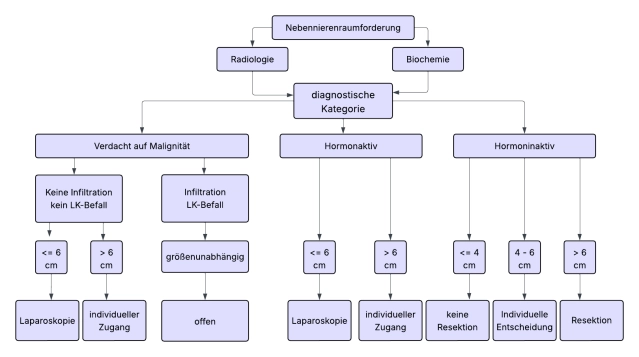
Há consenso nas diretrizes de que a maioria dos tumores adrenais deve ser operada de forma minimamente invasiva quando indicado. O diâmetro do tumor >6 cm e indicações claras de malignidade na imagem pré-operatória são considerados limites da cirurgia minimamente invasiva.
Para adrenalectomia aberta, a abordagem transabdominal ou toracoabdominal é preferida, pois a principal indicação para o procedimento aberto são tumores grandes suspeitos de malignidade.
Para tumores adrenais suspeitos de 6-8 cm de tamanho e unidades Hounsfield (UH) (valores de densidade da massa adrenal) >20, uma abordagem aberta (+linfadenectomia) é preferida sem evidência pré-operatória de malignidade.
Deve-se visar uma ressecção em bloco R0 com tecido adiposo retroperitoneal circundante, deve-se evitar a abertura da cápsula tumoral e, pelo menos, linfonodos morfologicamente conspícuos devem ser removidos simultaneamente.
Nas seguintes situações, pode existir uma indicação para adrenalectomia aberta:
- Tumores do córtex adrenal endócrinos ativos (adenomas de Conn ou Cushing, tumores com secreção de androgênios) especialmente com diâmetro > 10 cm
- Síndrome de Conn (hiperaldosteronismo primário, HPA):
A causa mais comum de hipertensão secundária é o hiperaldosteronismo primário.
No caso de um adenoma produtor de aldosterona unilateral, a adrenalectomia unilateral está indicada.
Em pacientes com hiperaldosteronismo primário (HPA) e alterações adrenais bilaterais, a adrenalectomia unilateral pode ser considerada se a amostragem venosa adrenal (AVA; coleta seletiva de sangue das veias adrenais) demonstrar clara lateralização funcional. O significado do CXCR4-PET como alternativa não invasiva à AVA deve ser validado por estudos adicionais.
- Síndrome de Cushing(hipercortisolismo)
Uma síndrome de Cushing adrenal florida com estigmas clínicos clássicos representa uma indicação para cirurgia.
Antes de uma possível remoção cirúrgica do tumor adrenal, deve-se confirmar a independência de ACTH do excesso de cortisol para evitar que o procedimento seja realizado erroneamente, embora a causa do excesso hormonal seja, por exemplo, hipofisária devido a um adenoma da hipófise anterior (síndrome de Cushing central) ou síndrome paraneoplásica com secreção ectópica de ACTH em doença tumoral.
- Tumores do córtex adrenal produtores de hormônios sexuais
Um carcinoma adrenocortical é a causa mais comum de secreção patológica clinicamente relevante de androgênios/estrogênios pela glândula adrenal, os adenomas são muito raros.
- Síndrome de Conn (hiperaldosteronismo primário, HPA):
- Carcinoma adrenocortical Carcinoma adrenocortical (CAC) estágio ENSAT I-III Link para classificação ENSAT
- No diagnóstico inicial, quase sempre > 4cm e exibe atividade endócrina em 50-80%. É típica a produção de cortisol ou produção hormonal mista (androgênios/estrogênios e cortisol).
- No carcinoma adrenocortical, a adrenalectomia aberta é o padrão ouro. Para tumores < 6 cm sem evidência de infiltração local ou linfonodal (Est. ENSAT I+II), a adrenalectomia minimamente invasiva pode ser realizada.
- Uma ressecção tumoral radical com remoção da glândula adrenal e todo o tecido adiposo/conectivo no compartimento afetado sem abertura da cápsula e uma linfadenectomia, esta com situação de dados incerta, é recomendada. Uma definição quanto à extensão da linfadenectomia necessária ainda não está disponível.
- Nota: A taxa de recidivas locais e peritoneais está aumentada no grupo laparoscópico de acordo com as evidências atuais. A conversão de adrenalectomia laparoscópica para aberta piora a sobrevida global.
- Feocromocitoma (FC) Tumor da medula adrenal com excesso de catecolaminas em casos de suspeita de malignidade, tumores muito grandes ou quando se esperam dificuldades técnicas.
- Aproximadamente 1/3 de todos os pacientes com feocromocitoma estão associados a uma síndrome tumoral hereditária. O rastreio genético é parte indispensável do diagnóstico de feocromocitoma. Os tumores associados a SDHB apresentam risco particularmente alto de malignidade e recidiva.
- No geral, 10% dos FCs são malignos. As metástases são a única prova de um feocromocitoma maligno.
- Mielolipomas não constituem per se uma indicação para cirurgia.
Grandes mielolipomas adrenais (tumores benignos hormonalmente inativos contendo tecido adiposo maduro e tecido hematopoiético) podem tornar-se sintomáticos por necrose ou sangramento espontâneo. Nesses casos, a ressecção pode ser necessária.
- Metástases adrenais (de tumores malignos de outras origens) devem ser removidas se não houver outras metástases presentes e a liberdade tumoral puder ser alcançada por meio da remoção.
Nota: A adrenalectomia por metástase deve ser realizada de forma minimamente invasiva, desde que a metástase possa ser removida in toto e sem disseminação de células tumorais. Uma abordagem aberta é reservada para os poucos casos em que há evidência de infiltração local ou se a metástase exceder 6 cm.
- Grandes schwannomas ou neuroblastomas em adultos