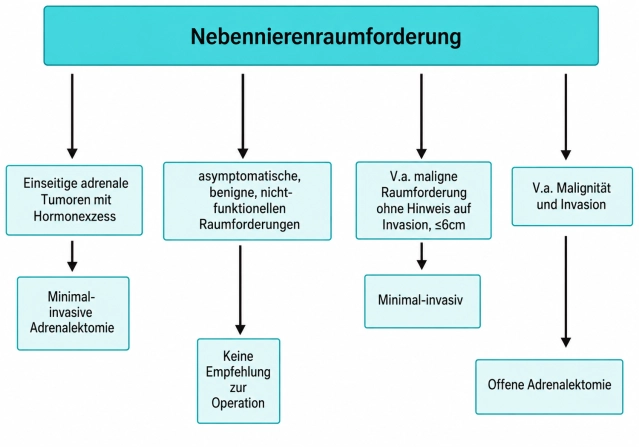
In den Leitlinien besteht Konsens, dass die meisten Nebennierentumoren (NN-Tumoren) bei gegebener Indikation minimal-invasiv operiert werden sollten. Tumordurchmesser > 6 cm und deutliche Hinweise auf Malignität in der präoperativen Bildgebung gelten als Grenzen der minimal-invasiven Chirurgie. Hinsichtlich der Tumorgröße wird vielfach von einer 6 cm-Regel ausgegangen, größere Tumore sollen konventionell-offen exstirpiert werden. Demgegenüber haben zahlreiche Untersuchungen gezeigt, dass auch bei großen Nebennierentumoren > 6 cm bei entsprechender Expertise sicher minimalinvasiv vorgegangen werden kann. Aufgrund fehlender Evidenz kann deshalb die 6 cm-Regel nicht als absolute Grenze angesehen werden. Dennoch wird generell bei großen Tumoren, insbesondere bei bildgebendem Malignitätsverdacht, die konventionelle Adrenalektomie empfohlen.
Laparoskopische und retroperitoneoskopische Verfahren sind gleichwertig. Die Wahl des Zugangswegs ist abhängig von der Erfahrung und der Präferenz des Operateurs.
Bemerkung: Die laparoskopische Adrenalektomie (AE) hat sich nach der Erstbeschreibung 1992 technisch nicht wesentlich verändert und ist wegen des vertrauten Zugangs und Orientierung vielerorts die bevorzugte Methode.
Die Nebennierenteilresektion (nebennierenerhaltende Adrenalektomie) hat einen Stellenwert bei Patienten mit Conn-Syndrom, das von kleinen meist exzentrisch liegenden Tumoren verursacht wird, und bilateralen Tumoren, bei denen der operative Eingriff mit dem Ziel des adrenokortikalen Funktionserhalts angestrebt wird. Hierbei soll der Erhalt von mindestens einem Drittel einer Nebenniere erreicht werden. Die zentrale Nebennierenvene muss nicht erhalten werden. Bei nur einseitiger Chirurgie wird die Kortikoidhormonproduktion in der Regel vollständig von der kontralateralen Nebenniere übernommen.
In folgenden Situationen ist eine Indikation zur minimal-invasiven Adrenalektomie gegeben:
- Endokrin-aktive Tumoren der Nebennierenrinde (Conn- oder Cushing-Adenome, Tumoren mit Sexualhormonsekretion) bis 10 cm
- Conn-Syndrom (primärer Hyperaldosteronismus, PHA):
Die häufigste Ursache einer sekundären Hypertonie ist der primäre Hyperaldosteronismus.
Screening bei Inzidentalom und gleichzeitiger Hypertonie und/oder unklarer Hypokaliämie. Morphologisch kann eine Hyperplasie der Nebennieren oder ein oder mehrere Adenome vorliegen.
Bei einem unilateralen aldosteronproduzierenden Adenom oder einer unilateralen adrenalen Hyperplasie ist die unilaterale Adrenalektomie indiziert. Beim solitären Aldosteron-produzierenden Adenom kann auch eine partielle Entfernung der betroffenen Nebenniere vorgenommen werden.
Bei Patienten mit primärem Hyperaldosteronismus (PHA) und bilateralen Nebennierenveränderungen kann eine einseitige Adrenalektomie in Betracht gezogen werden, sofern das adrenale Venensampling (AVS; selektive Blutentnahme aus den Nebennierenvenen) eine eindeutige funktionelle Lateralisierung nachweist. Die Bedeutung der CXCR4-PET als nicht-invasive Alternative zum AVS muss durch weitere Studien validiert werden.
- Cushing-Syndrom (Hypercortisolismus)
Ein florides adrenales Cushing-Syndrom mit klassischen klinischen Stigmata stellt eine OP-Indikation dar.
Eine besondere Situation liegt bei der milden autonomen Cortisolsekretion (MACS) vor. Definitionsgemäß weisen Patienten und Patientinnen mit MACS keine klinischen Zeichen eines Cushing-Syndroms auf, jedoch einen pathologischen 1-mg-Dexamethason-Hemmtest.
Eine besondere Situation liegt bei der milden autonomen Cortisolsekretion (MACS) vor. Definitionsgemäß weisen Patienten und Patientinnen mit MACS keine klinischen Zeichen eines Cushing-Syndroms auf, jedoch einen pathologischen 1-mg-Dexamethason-Hemmtest.
Eine große, 2022 publizierte Studie zeigte, dass Patienten und Patientinnen mit autonomer Cortisolsekretion im Vergleich zu Personen mit hormoninaktivem Adenom eine erhöhte Gesamtmortalität aufweisen (Deutschbein et al., Lancet Diabetes & Endocrinology, 2022). Gemäß der überarbeiteten Inzidentalom-Leitlinie der European Society of Endocrinology aus dem Jahr 2023 sollte bei MACS grundsätzlich eine operative Therapie erwogen werden, insbesondere bei Vorliegen relevanter Komorbiditäten.
Vor einer möglichen operativen Entfernung des NN-Tumors muss die ACTH-Unabhängigkeit des Kortisolexzesses bestätigt werden, damit der Eingriff nicht fälschlicherweise erfolgt, obwohl die Ursache des Hormonexzesses beispielsweise hypophysär bedingt ist durch ein Adenom des Hypophysenvorderlappens (zentrales Cushing-Syndrom) oder paraneoplastisches Syndrom mit ektoper ACTH-Sekretion bei Tumorerkrankung.
- Sexualhormon produzierende Nebennierenrindentumoren
Ein adrenokortikales Karzinom ist die häufigste Ursache für eine klinisch relevante, pathologische Androgen-/Östrogensekretion aus der Nebenniere, Adenome sind sehr selten.
- Conn-Syndrom (primärer Hyperaldosteronismus, PHA):
- Nebennierenrindenkarzinom Adrenokortikales Karzinom (ACC) bis zum ENSAT-Stadium II
Link zu ENSAT-Klassifikation und Größe ≤ 6cm- Bei der Erstdiagnose fast immer > 4cm und weist in 50-80% eine endokrine Aktivität auf. Typisch ist eine Cortisolproduktion oder eine gemischt-hormonelle Produktion (Androgene/Östrogene und Cortisol).
- Beim Nebennierenrindenkarzinom ist die offene Adrenalektomie der Goldstandard. Bei Tumoren < 6 cm ohne Hinweis auf lokale oder Lymphknoteninfiltration (ENSAT Std. I+II) kann eine minimal-invasive Adrenalektomie erfolgen.
- Eine radikale Tumorresektion mit Entfernung der Nebenniere und des gesamten Fett-/Bindegewebes im betroffenen Kompartiment ohne Kapseleröffnung mit Lymphadenektomie, diese bei unsicherer Datenlage, wird empfohlen. Eine Definition hinsichtlich der Ausdehnung der erforderlichen Lymphadenektomie liegt bisher nicht vor.
- Bemerkung: Die Rate an lokalen und peritonealen Rezidiven ist in der laparoskopischen Gruppe laut aktueller Evidenz erhöht. Eine Konversion von laparoskopischer zu offener Adrenalektomie verschlechtert das Gesamtüberleben.
- Phäochromozytom (PC) Nebennierenmarktumor mit Katecholaminexzess
- Ca 1/3 aller Phäochromozytom-Patienten sind einem hereditären Tumorsyndrom zuzuordnen. Das genetische Screening ist unverzichtbarer Bestandteil der Phäochromozytom Diagnostik.
- Zu den familiären syndromalen Formen gehören die multiple endokrine Neoplasie Typ 2 (MEN 2), das von-Hippel-Lindau-Syndrom (VHL), die Neurofibromatose Typ 1 (NF1) und die Keimbahnmutationen der Succinat-Dehydrogenase-Untereinheit B und D (SDHB und SDHD).
- Sie unterscheiden sich von den sporadischen Formen durch Erkrankungsalter, Tumorlokalisation und ihr Risiko zur malignen Entartung.
- Syndromassoziierte hereditäre Phäochromozytome haben eine niedrigere Malignitätsrate als sporadische Tumoren, treten aber oft multifokal/bilateral auf.
- Eine wichtige Ausnahme sind die SDHB-assoziierten Tumoren mit besonders hohem Malignitäts- und Rezidivrisiko.
- Organerhaltende Resektionen sollten bei geeigneter Indikation (hereditäre Tumoren, bilaterale Befunde, junge Patienten) bevorzugt werden, sofern dies onkologisch sicher möglich ist, um eine postoperative Nebenniereninsuffizienz zu vermeiden und die Notwendigkeit einer lebenslangen Steroidabhängigkeit zu reduzieren.
- Insgesamt sind 10 % der PC maligne. Beweisend für ein malignes Phäochromozytom sind ausschließlich Metastasen.
- Inzidentalome
| Größe des Inzidentaloms | Empfehlung |
|---|---|
| < 4 cm | Keine OP, wenn hormonell inaktiv und benigne Bildgebung |
| 4 – 6 cm | Individuelle Entscheidung („Graubereich“), Abwägung von Malignitätsrisiko, Bildgebung, Wachstum, Patientenwunsch, Re-CT/MRT in 6 - 12 Monaten |
| > 6 cm | OP empfohlen, auch bei hormonell inaktiven Tumoren wegen erhöhtem Karzinomrisiko (25%) |
Ein Nebenniereninzidentalom ist eine zufällige, meist asymptomatische Raumforderung (RF) der Nebenniere, die nicht aufgrund eines klinischen Verdachts auf eine endokrine Erkrankung entdeckt wurde. Meist gilt eine Größe ab 1 cm im Durchmesser als relevanter Befund.
Aufgrund der möglichen Malignität und hormonellen Aktivität sollte jedes Nebenniereninzidentalom ab einer Größe von 1cm leitliniengerecht mittels biochemischer und radiologischer Methoden abgeklärt werden.
Neben der Größe muss die Wahrscheinlichkeit eines malignen Tumors erfasst werden. Hierzu empfiehlt die Leitlinie an erster Stelle eine Computertomographie (CT) ohne Kontrastmittel mit Bestimmung der Hounsfield-Einheiten (HU) zur Dichtemessung der Tumoren. Bei einer homogenen Raumforderung mit einer Dichte von ≤ 10 HU ist keine weitere Bildgebung erforderlich. Bei kleinen homogenen Tumoren bis 4 cm Durchmesser und HU zwischen 11 und 20 soll eine zusätzliche Bildgebung erfolgen, z. B. durch eine Magnetresonanztomographie (MRT) mit „chemical-shift“, eine CT mit „wash-out“, oder einer Fluordesoxyglukose-Positronenemissionstomographie (s.u. Diagnostik). Tumoren, die größer sind, heterogen oder mit HU > 20 müssen in einer interdisziplinären Besprechung diskutiert werden. Es sollte dann entweder eine zügige zusätzliche Bildgebung erfolgen bzw. eine Operation evaluiert werden. Sollte nach erweiterter Bildgebung und interdisziplinärer Besprechung keine Indikation zur Operation gestellt werden, wird eine Verlaufsbildgebung in 6 bis 12 Monaten empfohlen.
- Nebennierenmetastasen (von malignen Tumoren anderen Ursprungs) sollten entfernt werden, wenn keine weiteren Metastasen vorliegen und durch die Entfernung Tumorfreiheit erreicht wird. Die Metastasen-Adrenalektomie wird minimal-invasiv vorgenommen, sofern die Metastase in toto und ohne Tumorzellaussaat entfernt werden kann. Ein offenes Vorgehen bleibt den wenigen Fällen vorbehalten, bei denen es Hinweise für eine lokale Infiltration gibt, oder wenn die Metastase 6 cm überschreitet.
- Myelolipome/Nebennierenzysten stellen keine Operationsindikation dar. Der Tumordurchmesser ist prognostisch unbedeutend, da es sich um gutartige Tumoren handelt. Flankenschmerzen bedingt durch die Größe des Tumors oder eine Einblutung im Retroperitoneum können selten eine Operationsindikation darstellen.
Lymphknotendissektion
Bei vergrößerten Lymphknoten soll eine lokoregionäre Lymphadenektomie durchgeführt werden. Die Datenlage bzgl. einer periadrenalen/renal-hilären Lymphadenektomie ist unbefriedigend.
